 400-166-8600400-166-8600
400-166-8600400-166-8600
帕金森病(Parkinson’s disease,PD)
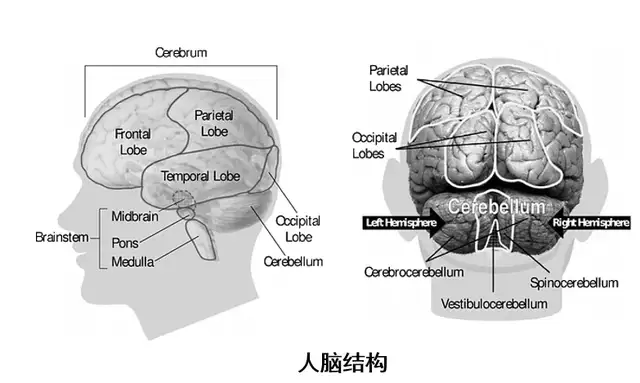
是一种常见的神经系统变性疾病,老年人多见,平均发病年龄为60岁左右,40岁以下起病的青年帕金森病较少见。我国65岁以上人群PD的患病率大约是1.7%。
大部分帕金森病患者为散发病例,仅有不到10%的患者有家族史。帕金森病最主要的病理改变是中脑黑质多巴胺(dopamine, DA)能神经元的变性死亡,由此而引起纹状体DA含量显著性减少而致病。
导致这一病理改变的确切病因仍不清楚,遗传因素、环境因素、年龄老化、氧化应激等均可能参与PD多巴胺能神经元的变性死亡过程。
病因
有研究表明,黑质(substantia nigra, SN)中多巴胺能神经元丧失会导致患者表现出的运动功能障碍。
PD致病因素多种多样,但有不少证据表明,线粒体功能缺陷是很重要的一项。例如:编码维持线粒体质量控制蛋白的PARK7、PARK6和PARK2基因突变能引起早发型PD。
由于多巴胺能神经元对代谢有很高的需求,因此造成其对线粒体功能障碍十分敏感。而线粒体氧化磷酸化(OXPHOS)的持续刺激,是以线粒体氧化损伤增加为代价的。
研究表明,PD患者SN中mtDNA完整性的丧失与功能性线粒体复合物I(MCI)的丧失存在一定的相关性。然而,这种MCI获得性损伤究竟是PD疾病进程中的一种副产品还是疾病的驱动因素还不得而知。
最新研究
为了探究线粒体-黑质-帕金森之间的关系,美国西北大学的研究团队进行了研究,最新结果发表在《Nature》上,论文标题为:“Disruption of mitochondrial complex I induces progressive parkinsonism"。

本文关键词
帕金森病(Parkinson’s disease,PD)
黑质(substantia nigra, SN)
多巴胺能神经元
功能性线粒体复合物I(MCI)
NDUFS4亚基
线粒体氧化磷酸化(OXPHOS)
浓缩就是精华版
使用交叉遗传学来破坏小鼠多巴胺能神经元的MCI功能。MCI的破坏诱导了代谢的warburg样改变,使神经元存活,但引发运动学习和精细运动缺陷。
因此,单是MCI功能障碍就足以引起进行性的类人帕金森病,在这种情况下,黑素多巴胺释放的缺失对运动功能障碍起关键作用。
并且人员还探究了不同类型的运动功能损伤,与不同部位多巴胺释放的相关性。
这些研究挑战了该疾病长期以来的主流的观点。
细胞研究离不开血清的支持,
点击下方小程序或结尾的“阅读原文"
了解更多血清产品
Ausbian进口特级胎牛血清
小程序
结果1:
采用Cre-loxP构建Ndufs2缺失小鼠品系。
得出最终结论:MCI的丧失会引起代谢重编程
大约在出生后第20天(P20)),条件下ndufs2敲除小鼠(cNdufs2−/−)的外观和大运动行为正常(MCI蛋白通常有20-40天的生命周期14,因此可能在出生后早期没有明显表型)。
在P20和P30之间,SN多巴胺能神经元中的线粒体成为三磷酸腺苷(ATP)的净消费者,而不是生产者,且线粒体嵴变化明显[这种变化在线粒体内膜(IMM)电位的敏感性中很明显——使用电位染料四甲基罗丹明(tetramethylrhodamine)对腺嘌呤核苷酸转运体(ANT)阻断的敏感性测量]。
cNdufs2−/−多巴胺能神经元中的体细胞线粒体密度是正常的。但是在许多cNdufs2−/−多巴胺能神经元中,线粒体嵴的结构发生了改变与电子传递链基因的下调一致。
分离野生型和cNdufs2−/−SN多巴胺能神经元的mRNA,并对其进行测序。在cNdufs2−/−多巴胺能神经元中存在大量的代谢重编程——一种类似warburg的效应。
促进糖酵解的基因编码蛋白上调,与OXPHOS相关的基因下调。编码糖酵解抑制因子的基因也被下调。
野生型多巴胺能神经元,寡霉素抑制线粒体复合物 V 导致 ATP 水平急剧下降。与之相比,在 cNdufs2−/− 多巴胺能神经元,该比率因糖酵解抑制而下降。

结果2:
除了触发代谢重编程外,Ndufs2的缺失还会导致轴突生长和转运相关基因(如Tubb3、Uchl1、Wnt5a、Sema3g、Nefl和Prkca)、突触传递相关基因(如Syt1、Syt3、Syt17、Syn2和Scna)的表达发生重大变化,DA合成/储存(如Th和Vamp2)和突触前调节(如Drd2, Chrna4和Chrna6)。背侧纹状体中酪氨酸羟化酶(TH)蛋白水平在P30左右下调,与线粒体OXPHOS的丧失平行。
对纹状体组织的液相色谱和质谱分析进一步验证cNdufs2-/-小鼠纹状体DA合成明显下降,此外,有助于驱动起搏的环核苷酸门控阳离子通道电流也明显减少。
结果3:
Ndufs2缺失后的体树突转移。
到P60,与多巴胺能信号相关的轴突蛋白的丢失由背侧纹状体扩大到腹侧纹状体。
cNdufs2-/-小鼠SN多巴胺能神经元胞体树突区域中的酪氨酸羟化酶表达降低至对照组一半左右。
DA释放量下降约75%。
组织学分析得出结论:在cNdufs2−/−小鼠病理进化的这个阶段,多巴胺能信号传递标记的丢失反映了表型下调,而不是明显的神经退行性变;也就是说,黑质纹状体轴突的分支形态是否保留仍有待确定。
对多巴胺能神经元进行标记后,发现cNdufs2−/−SN多巴胺能神经元的体树突递质表型的下降与对外部兴奋性刺激反应的损失并不紧密匹配。
结果4:
帕金森症的出现。
与在整个基底神经节中DA迅速耗尽的传统PD模型相比,cNdufs2-/-小鼠的病理分期能够评估DA释放的区域缺陷如何与行为相关联。
当背侧纹状体DA释放在P30左右下降到接近检测阈值时,cNdufs2−/−小鼠失去了执行联想学习任务的能力,而联想学习任务被认为依赖于DA依赖的纹状体突触可塑性。
在P30的全体性左旋多巴治疗(6 mg kg−1)可以恢复这一记忆任务,但在P60的后期治疗却不能。
在通过小鼠从前爪去除粘合剂所花费的时间来评估精细运动技能的实验中,cNdufs2-/-小鼠完成任务时间明显延长,同时也表现出较差的旷场探索行为表现。
P60的cNdufs2-/-小鼠仅表现出轻微的步态障碍,到了P100才会表现出后肢张开、爪子位置异常和步幅改变等特征。而在P120-150期间,大约有40%的SN多巴胺能神经元丢失。
结果5:
运动障碍的黑质决定簇。
分别向小鼠背侧纹状体或SN中立体定位注射携带AADC(可将左旋多巴转化为DA)的AAV,以及随后对小鼠旷场步态的分析,证明黑质多巴胺释放丧失对于粗大运动缺陷而言是必要因素。
★ 总结
这项研究,不仅证明多巴胺能神经元中MCI功能丧失足以引发进行性的、轴突先行的功能丧失和左旋多巴反应性帕金森病,还证明背侧纹状体的DA耗竭对于联想运动学习和精细动作而言是必要的,但黑质的DA释放缺陷才会引起类似于临床PD患者表现出的粗大运动损伤特征。
该模型不仅可以研究复合物 I 缺陷在疾病中的作用,还可以具有评估治疗策略的潜力。
参考文献:González-Rodríguez, Patricia et al. “Disruption of mitochondrial complex I induces progressive parkinsonism." Nature vol. 599,7886 (2021): 650-656.
doi:10.1038/s41586-021-04059-0
 当前位置:
当前位置: